天普大学Sudhir Kumar教授访问我室并做学术报告
2019年12月9日应陆剑研究员邀请,美国天普大学(Temple University) Sudhir Kmuar教授访问了北京大学蛋白质与植物基因重点实验室,并在金光生命科学楼邓祐才报告厅进行了题为“Molecular Evolution informs Genomic Medicine”的报告,讲座由陆剑研究员主持。
Sudhir Kumar教授任职于天普大学生物学系,并担任基因组学和进化医学研究所主任、Molecular Biology and Evolution杂志主编。其实验室致力于用分子进化和比较基因组学方法分析不同物种之间的亲缘关系和分歧时间,并将进化生物学方法应用于基因组医学研究,迄今发表170余篇文章,合计被引用超过17万次。其领导开发的MEGA软件累计引用已超15万次。
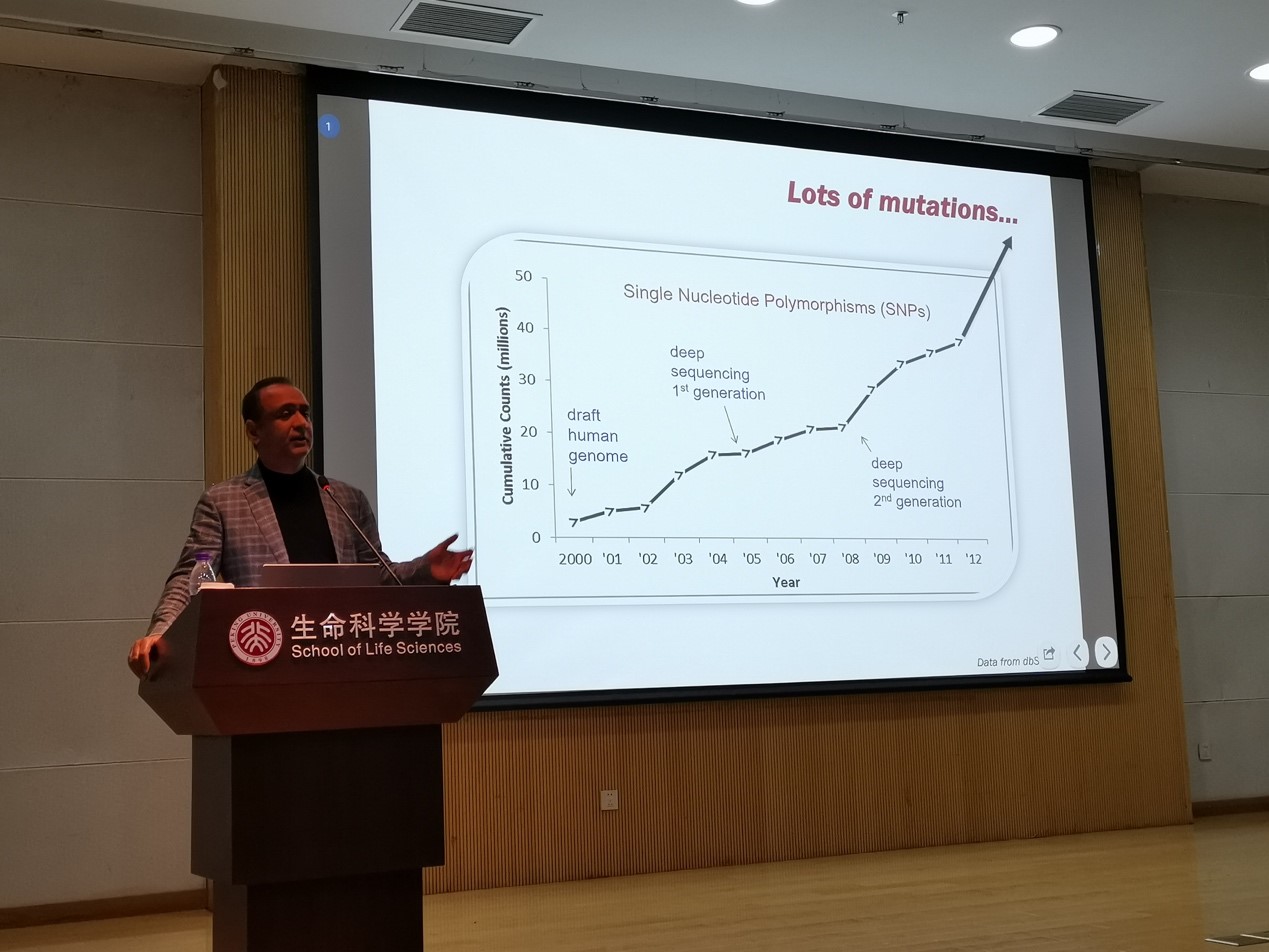
在当天的报告中, Kumar教授主要介绍了分子进化方法在基因组医学中的应用。人类外显子组中有大量的功能未知非同义单核苷酸变异(nSNV),预测这些nSNV对生物体的影响对解析人类遗传疾病至关重要。然而,当前已有的预测nSNV影响的软件包括Condel和polyphen-2的假阳性很高,尤其是在已知的富集疾病相关nSNV的超级保守位点。这是由于基因组中保守位点和不保守位点中的疾病相关突变的比例相差悬殊,这些方法将保守性不同的位点放在一起训练预测模型时很容易高估保守位点中疾病相关位点的比例。为此其课题组开发了EvoD算法,首先将nSNV根据所在位点的保守性分成不同组,在每组中分别考虑nSNV所在位点的进化速率和保守性以及所编码的氨基酸的化学性质来训练预测模型。与之前的方法相比,EvoD算法降低了疾病相关nSNV预测的假阳性率,并且在保守性不同的位点有相似的准确率。然而,EvoD算法仍然需要群体中的多态性数据以及已知的训练数据,然而很多时候我们很难获得理想的训练数据,并且一些低频的疾病相关突变可能很难在小群体中被检测到,为了解决这一问题,他们开发了不依赖于群体多态性数据和已知训练数据集的新算法。这一方法基于多物种氨基酸序列的比对,利用贝叶斯算法计算人类蛋白质序列中一个位点的不同氨基酸的进化概率(EP):一个等位基因所编码的氨基酸EP越低,即演化过程中越不可能出现,这个等位基因越有可能会导致人类疾病。EP可以用来区分中性的和与疾病相关的等位基因,跟其它需要更多数据的软件预测的结果有很高的一致性。另一方面,如果一个nSNV对应的氨基酸在演化过程中出现的概率很低 (EP<0.05)但在人类群体中的出现的频率反而较高(>0.05),这样的位点可能是受到正选择的位点。基于此,他们在人类群体中鉴定了超过18,000个Candidate Adaptive Polymorphism (CAP)位点,并通过多种方法排除了其它的可能解释,估计这其中至少有一半以上是真的适应性的位点。这些研究展现了如何用进化生物学方法对疾病相关的突变进行预测,为我们进行进化生物学研究并将发现与医学应用结合起来提供了范例。
Kumar教授还介绍了Molecular Biology and Evolution杂志的发展历史和审稿流程,并鼓励中国学者踊跃向该杂志投稿。
- 美国国家医学院院士Gerd A. Blobe受邀作学术报告2026.07.08
- 美国国家医学院院士GerdA.Blobe受邀作学术报告2026.07.08
- 上海交通大学农业与生物学院杨仲南教授应邀作学术报告2026.06.29
- 宾夕法尼亚州立大学生物系马红教授应邀作学术报告2026.06.26
- 中国科学院遗传与发育生物学研究所许操研究员应邀作学术报告2026.06.15
- 西安交通大学叶凯教授等人受邀作学术报告2026.05.29
- 德国马普所George Coupland院士受邀作学术报告2026.05.21
- 北京生命科学研究所苏俊研究员应邀作学术报告2026.05.20
- 郑州大学第一附属医院杨静华教授应邀访问并作学术报告2026.05.06
- 中国科学院院士、著名水稻遗传育种专家张启发教授应邀访问全国重点实验室并作学术报告2026.05.06
- 中国药科大学生物医药卓越工程师学院郭炜教授应邀访问并作学术报告2026.05.06
- 清华大学生命科学学院刘念副教授应邀作学术报告2026.04.29
- 斯坦福大学博士后王瀚宸应邀作学术报告2026.04.24
- 匹兹堡大学朱伯开教授应邀访问并作学术报告2026.04.18
- 哈佛医学院讲席教授Frederick W. Alt应邀访问并作学术报告2026.04.18
- Pablo Manavella 研究员应邀访问并作学术交流2026.04.17
- Don Cleveland教授应邀在金光生命科学大楼进行报告2026.04.16
- Ari Pekka Mähönen教授应邀访问并作学术交流2026.03.30
- 中国科学院化学研究所汪铭研究员应邀访问并作学术报告2026.03.25
- John Rasko教授应邀访问并作学术报告2026.03.10
- 中国科学院分子植物科学卓越创新中心杨卫兵研究员应邀访问并作学术报告2026.01.09
- 北京大学物理学院博雅特聘教授马滟青应邀访问并作学术报告2026.01.06
- 英国普利茅斯大学的Gert-Jan Jeunen教授应邀访问并作学术报告2025.12.30
- 华东师范大学生命科学学院翁杰敏教授应邀访问并作学术报告2025.12.19
- 哈佛大学医学院、麻省理工学院&哈佛大学博德研究所夏波研究员应邀访问并作学术报告2025.12.09
- 荷兰瓦赫宁根大学Dolf Weijers教授应邀访问并作学术报告2025.12.05
- 兰州大学杨晓燕教授应邀作学术报告2025.12.04
- 卡内基科学研究所王志勇博士应邀访问并作学术报告2025.12.02
- Jiří Friml教授应邀作学术交流2025.11.28
- 贝勒医学院RICHARD H. FINNELL教授应邀作学术报告2025.11.21
- 美国国家科学院院士Stephen J. O’Brien教授受邀来访并作学术报告2025.11.03
- Mark Stoneking教授应邀来访并作学术报告2025.10.27
- 哥伦比亚大学医学中心 Shan Zha教授应邀访问并作学术报告2025.10.17
- 奥地利分子病理学研究所教授与多伦多大学教授应邀访问并作学术报告2025.10.17
- 格雷戈尔·孟德尔分子植物生物学研究所Magnus Nordborg博士应邀访问并作学术报告2025.10.16
- 剑桥大学Aram Gurzadyan博士应邀来访并作学术交流2025.09.30
- 德国马克斯-普朗克研究所Diethard Tautz教授应邀作学术报告2025.08.02
- 匹兹堡大学医学院邢建华教授应邀访问并作学术报告2025.07.30
- 芝加哥大学杰出讲席教授龙漫远博士应邀访问全国重点实验室并作学术报告2025.07.26
- 圣裘德儿童研究医院Jiyuan Yang博士应邀学术访问并作报告2025.07.08
- 中国科学院分子植物科学卓越创新中心徐麟研究员应邀作学术交流2025.06.23
- 印第安纳大学韩冷教授应邀作学术报告2025.06.23
- 瑞士洛桑大学Niko Geldner教授应邀作学术报告2025.06.16
- 北京大学肿瘤医院吴健民研究员应邀作学术报告2025.06.16
- 中国药科大学廉腾飞教授应邀访问并作学术报告2025.05.21
- 法国国家科学研究中心Pierre Taberlet教授应邀来访并作学术报告2025.05.15
- 美国加利福尼亚大学戴维斯分校Siobhan M. Brady教授应邀访问并作学术报告2025.05.13
- 中国科学院分子植物学卓越创新中心赵杨研究员应邀访问全国重点实验室并作学术报告2025.05.09
- UC San Diego郝楠教授应邀学术访问2025.04.25
- 英国邓迪大学David M. J. Lilley 教授应邀访问并作学术报告2025.04.03
- 中国科学院昆明动物研究所王国栋研究员应邀学术访问并作报告2025.04.03
- 美国哥伦比亚大学教授丁磊应邀访问并作学术报告2025.03.25
- 罗格斯大学教授、上海交通大学特聘教授赵立平应邀访问并作学术报告2025.03.14
- 美国杜克大学教授、霍华德·休斯医学研究所研究员何胜洋院士应邀请访问并作学术报告2025.03.03
- 福建农林大学廖红教授应邀访问并作学术报告2025.01.16
- 华中农业大学李霞教授应邀访问并作学术报告2025.01.16
- 南京农业大学宋庆鑫教授应邀访问并作学术报告2025.01.16
- 哥本哈根大学张超助理教授应邀访问并作学术报告2024.12.25
- 军事科学院军事医学研究院张令强教授应邀访问并作学术报告2024.12.24
- 广州大学生命科学学院教授孔凡江博士应邀学术访问并作报告2024.11.20
- Dinshaw Patel教授受邀做学术报告2024.11.19
- 芝加哥大学教授Tao Pan应邀并做专题学术报告会2024.11.18
- 加州大学河滨分校教授Susan Wessler应邀学术访问并作报告2024.11.08
- 秋田县立大学Hiroo Fukuda教授应邀访问北京大学蛋白质与植物基因研究国家重点实验室并作学术报告2024.10.31
- Michael Gnant教授应邀访问国家重点实验室并做学术报告2024.10.31
- Marek Tchorzewski教授应邀访问国家重点实验室并进行报告2024.10.31
- Peter Kristensen教授应邀在吕志和大楼进行报告2024.09.14
- 德国维尔茨堡大学丁美琪博士应邀做学术报告2024.09.09
- 赫尔辛基大学Yrjö Helariutta教授应邀访问国家重点实验室并作学术报告2024.09.04
- 美国密歇根大学林建谍教授应邀请访问并做学术报告2024.07.17
- 浙江大学James Whelan教授应邀访问北京大学蛋白质与植物基因研究国家重点实验室并作学术报告2024.07.08
- 须健教授应邀来北京大学蛋白质与植物基因研究国家重点实验室做学术交流2024.07.08
- 芝加哥大学龙漫远教授访问北京大学蛋白质与植物基因研究国家重点实验室2024.07.06
- 中科院上海免疫与感染研究所Daniel Falush研究员应邀学术访问并作报告2024.06.26
- 清华大学基础医学院院长李海涛教授应邀请访问并做学术报告2024.06.11
- 复旦大学附属肿瘤医院、复旦大学生物医学研究院徐彦辉教授应邀请访问并做学术报告2024.05.31
- 上海交通大学农业与生物学院长聘教轨副教授高起飞博士应邀做学术报告2024.05.29
- 中国科学院分子细胞科学卓越创新中心周金秋研究员受邀做学术报告2024.05.22
- 中山大学李旭日教授应邀做学术报告2024.05.15
- 清华大学生命科学学院戚益军教授应邀做学术报告2024.04.25
- 美国威斯康星大学David Drake教授应邀访问重点实验室并做学术报告2024.04.24
- 耶鲁大学医学院药理学系助理教授米伟博士应邀做学术报告2024.04.15
- 理海大学任元必教授应邀做学术报告2024.04.15
- 德国科隆大学Siegfried Roth教授访问我国家重点实验室并做学术报告2024.04.03
- 四川大学生命科学学院张阳教授应邀做学术报告2024.04.01
- “RNA修饰网络的动态构筑与调节机制”专题学术报告会2024.04.01
- 英国皇家科学院院士Noni(V.E.) Franklin-Tong教授学术交流活动2024.03.28
- 中国科学院分子植物科学卓越创新中心王二涛研究员应邀访问国家重点实验室并作学术报告2024.03.13
- 同济大学生命科学与技术学院高绍荣教授应邀做学术报告2024.03.06
- 中国工程院院士张改平应邀做学术报告2024.03.04
- 澳大利亚莫纳什大学宋江宁教授应邀做学术报告2023.12.20
- 西湖大学生命科学学院的柴继杰教授应邀做学术报告2023.12.13
- 西湖大学生命科学学院管坤良教授应邀做学术报告2023.12.06
- 同济大学医学院王平教授应邀做学术报告2023.11.20
- 韩国蔚山国立科学技术研究所Kyungjae (KJ) Myung教授应邀学术访问2023.11.14
- 乌得勒支大学Friedrich Förster教授受邀访问北京大学并做学术报告2023.11.09
- 上海交通大学乔中东教授应邀来我国家重点实验室作学术交流2023.11.07
- 中国科学院院士邓子新教授应邀来我国家重点实验室作学术交流2023.11.07
- 中科院冯献忠研究员受邀前往北大生科院作学术报告2023.11.06
- 英国塞恩斯伯里研究所马文勃教授受邀访问并做学术报告2023.10.26